New molecular insights into a rare neurological disorder
Investigations by the Sondermann group (DESLY, CAU) into the molecular mechanisms of the dynamin-related protein Atlastin (ATL) have revealed important insights that expand our understanding of a group of neurogenerative disorders.
Hereditary spastic paraplegia (HSP) is a rare group of genetic disorders affecting roughly one in every 11,000 individuals. Those suffering from HSP typically experience a weakness and stiffness in the legs which worsens over time and approximately 10% of individuals experiences a more complex form of the disorder, which includes other neurological symptoms.
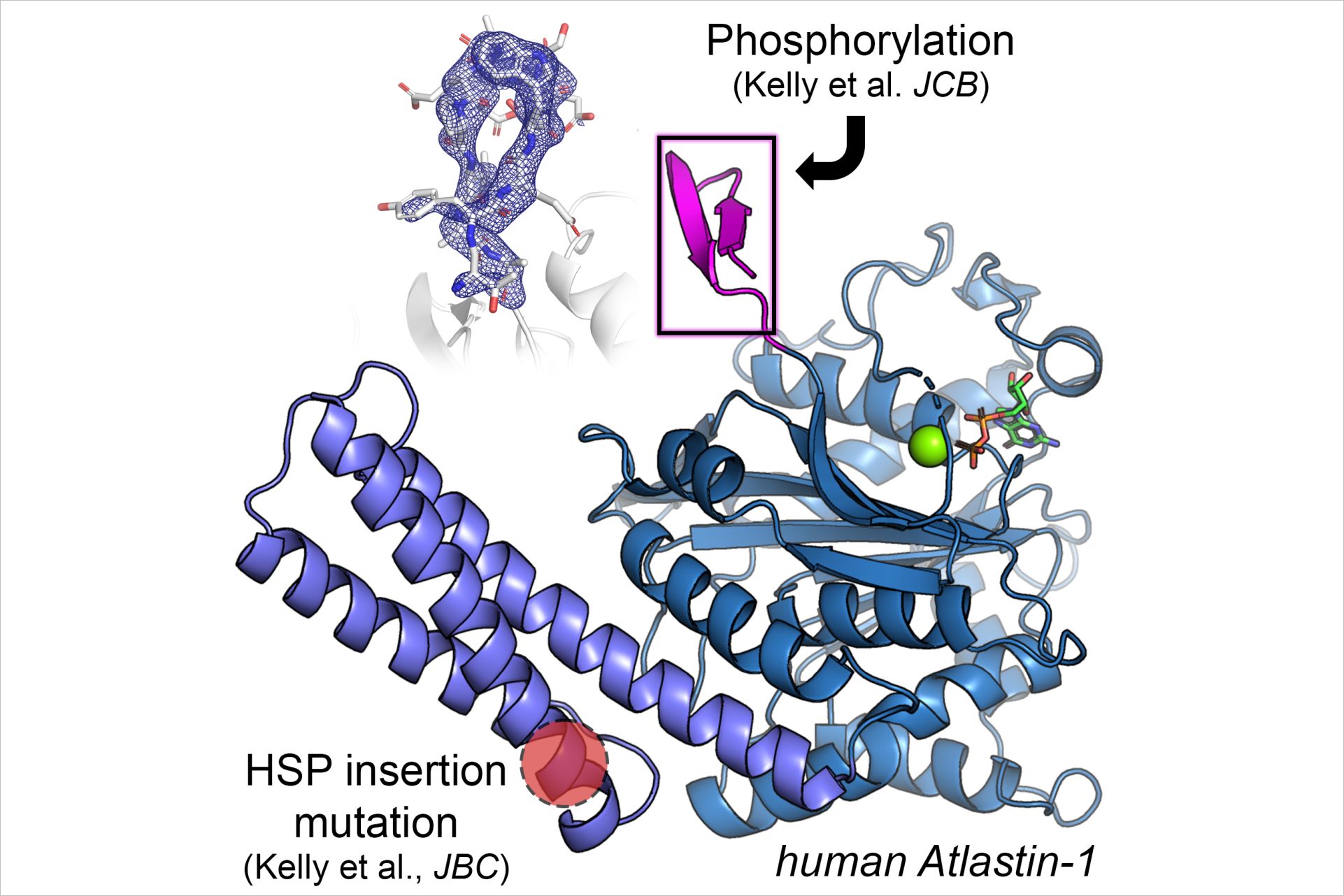
HSP is caused by mutations in individual genes that have been inherited by one or both parents. Mutations in the gene ATL1 account for approximately 10% of all HSP cases and 80% of early-onset cases. In eukaryotic cells, ATLs are responsible for the characteristic branching of the smooth endoplasmic reticulum, an interconnected network of membrane-enclosed tubules. In a study recently published in the Journal of Cell Biology, the researchers revealed, using structural and functional approaches, that a short sequence motif that distinguishes ATL isoforms, the N-terminal hypervariable region of (HVR), is a site for regulation of ATL activity. “Understanding more about how ATL1 functions enables us to identify cellular effectors of ATL-mediated membrane processes,” explains Sondermann.
In another study published in the Journal of Biological Chemistry, the researchers investigated a newly discovered ATL1 mutation in a patient with complex HSP. The mutation, an insertion of three nucleotides, which translates into an additional asparagine residue in the protein sequence, affects a central domain of ATL1. Examining the molecular structure of the ALT1 gene product with the mutation revealed a noticeable conformation alteration in the protein domain. Although the basic activity of the enzyme was unaffected by the mutation, the mutant protein was more efficient in tethering membranes than the unaltered, wild-type version. “Typically, ATL1 mutations cause a loss of the enzyme’s function,” notes Sondermann “we were surprised to see that this specific insertion mutation was not only tolerated, resulting in folded, active protein; but it showed gain-of-function characteristics as a potential disease mechanism, which is uncommon for ATL1-associated HSPs.”
While these findings will not alter the course of HSP for this individual patient, the continued discovery and characterization of novel disease mutations is crucial for our overall understanding of how HSP develops. The understanding of ALT1s molecular mechanisms gained in these two studies will contribute to our basic understanding of the regulation of these proteins and the diagnostics for this debilitating disease.
The work was supported by the Spastic Paraplegia Foundation. Lead author on both studies is Carolyn M. Kelly, a former graduate student in the Sondermann group. The work on the novel ATL1 mutation is based on a close collaboration with Keri Ramsey and the Center for Rare Childhood Disorders, Translational Genomics Research Institute (TGen) in Phoenix, Arizona, USA.
Sources:
Kelly CM, Byrnes LJ, Neela N, Sondermann H, O'Donnell JP (2021) The hypervariable region of atlastin-1 is a site for intrinsic and extrinsic regulation. J Cell Biol. 220(11):e202104128. doi: 10.1083/jcb.202104128
Kelly CM, Zeiger PJ, Narayanan V, Ramsey K, Sondermann H (2021) A novel insertion mutation in Atlastin 1 is associated with spastic quadriplegia, increased membrane tethering, and aberrant conformational switching. J Biol Chem. 101438. doi: 10.1016/j.jbc.2021.101438